Documentation
The first section of this documentation explains how to download, install and run LotuS2 in no time. However, for a more in-depth understanding it is recommended to read the full documentation, familiarize yourself with the basic commandline options and follow the LotuS2 tutorial (currently under revision) and the Numerical Ecology tutorial with R. This should set you up to conduct your own analyses with LotuS2. If you struggle and can not find the solution in the documentation, have a look at the FAQs or contact me (Contact).
Installation
To use LotuS2 you need to either install it manually using github, or install it via conda or docker. If you opt for conda or docker you can skip to Setup.
Note on usearch/uparse
Due to licence restrictions, usearch (download usearch) has to be downloaded and installed by the users, all other programs are automatically installed and configured by executing the autoInstall.pl. All proprietary software used in your specific LotuS2 run will be listed, including the relevant citation, in "output_dir/citations.txt". Please refere to the SILVA Terms of Use/License Information".
Downloads
For installing LotuS2 manually, you have to download the latest release from github.
Clone the LotuS2 repository to download the latest github version:Manual setup
LotuS2 comes with an autoinstaller script to automatically set up LotuS2 and download the required databases. This step is only necessary when installing LotuS2 from github. Change directory into the previously extracted lotus folder and run the following command to start the autoinstall process. For the autoinstall no sudo permissions are required. The autoinstall script will automatically guide you through the installation.
./autoInstall.pl
If usearch is not already installed and in your path, autoinstall will ask you to specify an absolute path to the usearch binary. You can skip this step and setup usearch later with one of the following commands:
./autoInstall.pl -link_usearch [path to usearch]
# link usearch temporarily when calling lotus2 with the flag -link_usearch [path]
./lotus2 -link_usearch [path to usearch] ...
Setup with conda
LotuS2 can be installed via conda. For that you have to first add the channels bioconda and conda-forge. As LotuS2 has many dependencies, it is advised to install the program in a separate conda environment (as stated in the following).

conda config --add channels conda-forge
conda create -n lotus2 lotus2
conda activate lotus2
Setup with docker
The docker image can be found on the download page. A detailed description for docker can be found here.
Update LotuS2
LotuS2 is frequently updated to improve user experience and to include new functionalities. To update LotuS2 after a manual installation, follow the next steps to get the latest version from github. After an update with "git pull" the databases are still there and there is no need to run autoinstall again.
cd lotus2
# pull latest version
git pull
You can also perform a fresh install, but then you have to rerun the autoinstall script again.
Important: The latest version on Github via "git pull" might not be same as the latest conda version.
With conda, you only need to run:
conda activate lotus2
# run update command
conda update lotus2
Troubleshooting
Renaming or relocating LotuS2 installation:
Please be aware that if you relocate the LotuS2 folder LotuS2 will fail to find the utilized third-party software. This is because LotuS2 stores absolute paths to the programs in the lOTUs.cfg file. If you want to move your folder you can change the paths in the config file with the following command:
Important for dada2:
If you would like to use dada2, LotuS2 will attempt to install this via autoInstall.pl. However, in our experience this is instable and we recommend to install dada2 manually in your global R distribution first, before starting the LotuS2 install. LotuS2 will require Rscript accessible in your bash.
Lotus2 Usage
LotuS2 is a perl script that links several programs to demultiplex and cluster 16S amplicon sequences. The general syntax to LotuS2 is:
Basic Options
-i <file|dir>
In case that fastqFile or fnaFile and qualFile were specified in the mapping file, this has to be the directory with input files
-o <dir>
Warning: The output directory will be completely removed at the beginning of the LotuS2 run. Please ensure this is a new directory or contains only disposable data!
-m|-map <file>
Mapping file
Further Options
-q <file>
.qual file associated to fasta file. This is an old format that was replaced by fastq format and is rarely used nowadays. (Default: "")
-barcode|-MID <file>
Filepath to fastq formated file with barcodes (this is a processed mi/hiSeq format). The complementary option in a mapping file would be the column "MIDfqFile". (Default: "")
-s|sdmopt <file>
SDM option file, defaults to "configs/sdm_miSeq.txt" in current dir. (Default: miSeq)
-c <file>
LotuS.cfg, config file with program paths. (Default: <LotuS2_dir>/lOTUs.cfg)
-p <454/miSeq/hiSeq/PacBio>
sequencing platform: PacBio, 454, miSeq or hiSeq. (Default: miSeq)
-t|-threads <num>
number of threads to be used. (Default: 1)
-tmp|-tmpDir <dir>
temporary directory used to save intermediate results. (Default: <outputDir>/tmpDir)
Workflow Options
-forwardPrimer <string>
give the forward primer used to amplify DNA region (e.g. 16S primer fwd)
-reversePrimer <string>
give the reverse primer used to amplify DNA region (e.g. 16S primer rev)
-verbosity <0-3>
Level of verbosity from printing all program calls and program output (3) to not even printing errors (0). (Default: 1)
-backmap_id <0-1>
%id cutoff for backmapping mid-qual reads onto OTUs/zOTUs/ASVs (Default: 0.97 or 0.99 for ASVs/zOTUs)
-saveDemultiplex <0|1|2|3>
(1) Saves all demultiplexed reads (unfiltered) in the [outputdir]/demultiplexed folder, for easier data upload. (2) Only saves quality filtered demultiplexed reads and continues LotuS2 run subsequently. (3) Saves demultiplexed, filtered reads into a single fq, with sample ID in fastq/a header. (0) No demultiplexed reads are saved. (Default: 0)
-taxOnly <file>
Skip most of the lotus pipeline and only run a taxonomic classification on a fasta file. E.g. lotus2 -taxOnly <some16S.fna> -refDB SLV
-redoTaxOnly <0|1>
(1) Only redo the taxonomic assignments (useful for replacing a DB used on a finished lotus run). (0) Normal lotus run. (Default: 0)
-offtargetDB <file>
Remove likely contaminant OTUs/ASVs based on alignment to provided fasta. This option is useful for low-bacterial biomass samples, to remove possible host genome contaminations (e.g. human/mouse genome)
-keepOfftargets <0|1>
(0)?!?: keep offtarget hits against offtargetDB in output fasta and otu matrix. (Default 0)
-keepTmpFiles <0|1>
(1) save extra tmp files like chimeric OTUs or the raw blast output in extra dir. (0) do not save these. (Default: 0)
-keepUnclassified <0|1>
(1) Includes unclassified OTUs (no Phylum assignment) in OTU and taxa abundance matrix calculations. (0) does not report these potential contaminants. (Default: 1)
-intargetDB <file>
Keep all OTUs with good matches to this DB (.fa format). This option is useful if you have a set of known true positive 16S sequences, that might not be represented in your tax DB and would otherwise be removed through \"-keepUnclassified 1\".
-tolerateCorruptFq <0|1>
(1) Continue reading fastq files, even if single entries are incomplete (e.g. half of qual values missing). (0) Abort lotus run, if fastq file is corrupt. (Default: 0)
-useVsearch <0|1>
(0) Use usearch for internal tasks such as remapping reads on OTUs, chimera checks. (1) will use vsearch for these tasks. This option is independent of the -CL UPARSE/UNOISE option, and -taxAligner assignment usearch/vsearch options. (Default: 0)
-mergePreClusterReads <0|1>
(0) no merging or reads pre OTU/ASV/zOTU seq clustering, BUT read merging after seq clustering (to get better representative sequence). (1) Merge reads prior to seq clustering. WARNING!! This will considerably reduce the number of valid read pairs, as additional quality filters will be applied, algorithm is still in development !! (Default: 0)
Taxonomy Options
-taxAligner <0|blast|lambda|utax|sintax|vsearch|usearch>
(0) alginment deactivated, use RDPclassifier (this does not report species level taxonomies); (1) or (blast) use Blast; (2) or (lambda) use LAMBDA to search against a 16S reference database for taxonomic profiling of OTUs; (3) or (utax) or (sintax): use UTAX/SINTAX with custom databases; will use SINTAX if uparse ver >= 9 is found (4) or (vsearch) use VSEARCH to align OTUs to custom databases; (5) or (usearch) use USEARCH to align OTUs to custom databases. (Default: 0)
-refDB <KSGP|SLV|GG2|HITdb|PR2|UNITE|beetax>
(SLV) Silva LSU (23/28S) or SSU (16/18S), (KSGP) Bacteria, Archaea, Eukaryotes SSU, (GG2) GreenGenes2 SSU, (HITdb) human gut specific SSU, (PR2) LSU spezialized on Ocean environmentas, (UNITE) ITS fungi specific, (beetax) bee gut specific SSU. Given that \"-amplicon_type \" was set to SSU or LSU, the appropriate DB in SLV would be used. \nDecide which reference DB will be used for a similarity based taxonomy annotation. Databases can be combined, with the first having the highest priority. E.g. "HITdb,SLV" would priority assign OTUs to PR2 taxonomy, but hits with a higher %id to SLV would be assigned to SLV. Can also be a custom fasta formatted database: in this case provide the path to the fasta file as well as the path to the taxonomy for the sequences using -tax4refDB. For custom databases QIIME2 file formats are compatible if the delimiter in the QIIME2 taxonomy file is changed from semicolon to tab. See also online help on how to create a custom DB. WARNING: combining databases with incompatible tax levels (e.g. PR2,SLV) will result in non sensical tax levels. (Default: none)
-tax4refDB <file>
In conjunction with a custom fasta file provided to argument -refDB, this file contains for each fasta entry in the reference DB a taxonomic annotation string, with the same number of taxonomic levels for each, tab separated.
-amplicon_type <SSU|LSU|ITS|ITS1|ITS2|custom>
(SSU) small subunit (16S/18S), (LSU) large subunit (23S/28S) or internal transcribed spacer (ITS|ITS1|ITS2), (custom) for custom marker genes. These options will change default read qual filter parameters and activate ITS specific postfiltering steps. (Default: SSU)
-tax_group <bacteria|fungi|eukarya>
(bacteria) bacterial 16S rDNA annnotation, (fungi) fungal 18S/23S/ITS annotation, (eukarya) eukaryotic (18S/23S) annotation. (Default: bacteria)
-rdp_thr <0-1>
Confidence thresshold for RDP. (Default: 0.8)
-sintax_thr <0-1>
Confidence thresshold for SINTAX. (Default: 0.8)
-useBestBlastHitOnly <0|1>
(1) do not use LCA (lowest common ancestor) to determine most likely taxonomic level (not recommended), instead just use the best blast hit. (0) LCA algorithm. (Default: 0)
-LCA_cover <0-1>
Min horizontal coverage of an OTU sequence against ref DB. (Default: 0.5)
-LCA_frac <0-1>
Min fraction of database hits at taxlevel, with identical taxonomy. (Default: 0.9)
-LCA_idthresh <97,95,93,91,88,78>
6 numbers, comma separated, that are min %id of OTU/ASV fasta to ref database, to assign taxonomy to OTU/ASV at this taxonomic level
-taxExcludeGrep <string>
Exclude taxonomic group, these OTUs will be assigned as unknown instead. E.g. -taxExcludeGrep Chloroplast|Mitochondria (Default: )
-greengenesSpecies <0|1>
(1) Create greengenes output labels instead of OTU (to be used with greengenes specific programs such as BugBase). (Default: 0)
-ITSx <0|1>
(1) use ITSx to only retain OTUs fitting to ITS1/ITS2 hmm models; (0) deactivate. (Default: 1)
-itsx_partial <0-100>
Parameters for ITSx to extract partial (%) ITS regions as well. (0) deactivate. (Default: 0)
-lulu <0|1>
(1) use LULU (https://github.com/tobiasgf/lulu) to merge OTUs based on their occurrence. (Default: 1)
-buildPhylo <0,1,2,>
(0) do not build OTU phylogeny; (1) use fasttree2; (2) use IQ-TREE 2. (Default: 1). We recommend the cautious usage of the phylogenetic tree for ITS because high variation of ITS sequences may lead to erroneous trees. Phylogenetic trees can be of use for 16S data depending on the aim of the analysis.
Clustering Options
-CL|-clustering <uparse|swarm|cdhit|unoise|dada2|vsearch>
Sequence clustering algorithm: (1) UPARSE, (2) swarm, (3) cd-hit, (6) unoise3, (7) dada2, (8) VSEARCH. Short keyword or number can be used to indicate clustering (Default: UPARSE)
-id <0-1>
Clustering threshold for OTUs. (Default: 0.97)
-swarm_distance <1,2,3,..>
Clustering distance for OTUs when using swarm clustering. (Default: 1)
-chim_skew <num>
Skew in chimeric fragment abundance (uchime option). (Default: 2)
-count_chimeras<T/F>
Add chimeras to count up OTUs/ASVs. (Default: F)
-deactivateChimeraCheck <0|1|2|3>
(0) do OTU chimera checks. (1) no chimera check at all. (2) Deactivate deNovo chimera check. (3) Deactivate ref based chimera check. (Default: 0)
-derepMin<num>
Minimum size of dereplicated clustered, one form of noise removal. Can also have a more complex syntax, see examples. (Default: 8:1,4:2,3:3)
-readOverlap <num>
The maximum number of basepairs that two reads are overlapping. (Default: 300)
-endRem <string>
DNA sequence, usually reverse primer or reverse adaptor; all sequence beyond this point will be removed from OTUs. This is redundant with the "ReversePrimer" option from the mapping file, but gives more control (e.g. there is a problem with adaptors in the OTU output). (Default: "")
-xtalk <0|1>
(1) check for crosstalk. Note that this requires in most cases 64bit usearch. (Default: 0)
Other uses of pipeline (quits after execution)
-v
Print LotuS2 version
-link_usearch <file>
Provide the absolute path to your local usearch binary file, this will be installed to be useable with LotuS2 in the future.
-check_map <file>
Mapping_file: only checks mapping file and exists.
-create_map <file>
mapping_file: creates a new mapping file at location, based on already demultiplexed input (-i) dir. E.g. lotus2 -create_map mymap.txt -i /home/dir_with_demultiplex_fastq
-taxOnly <file>
Skip most of the lotus pipeline and only run a taxonomic classification on a fasta file. E.g. lotus2 -taxOnly <some16S.fna> -refDB SLV
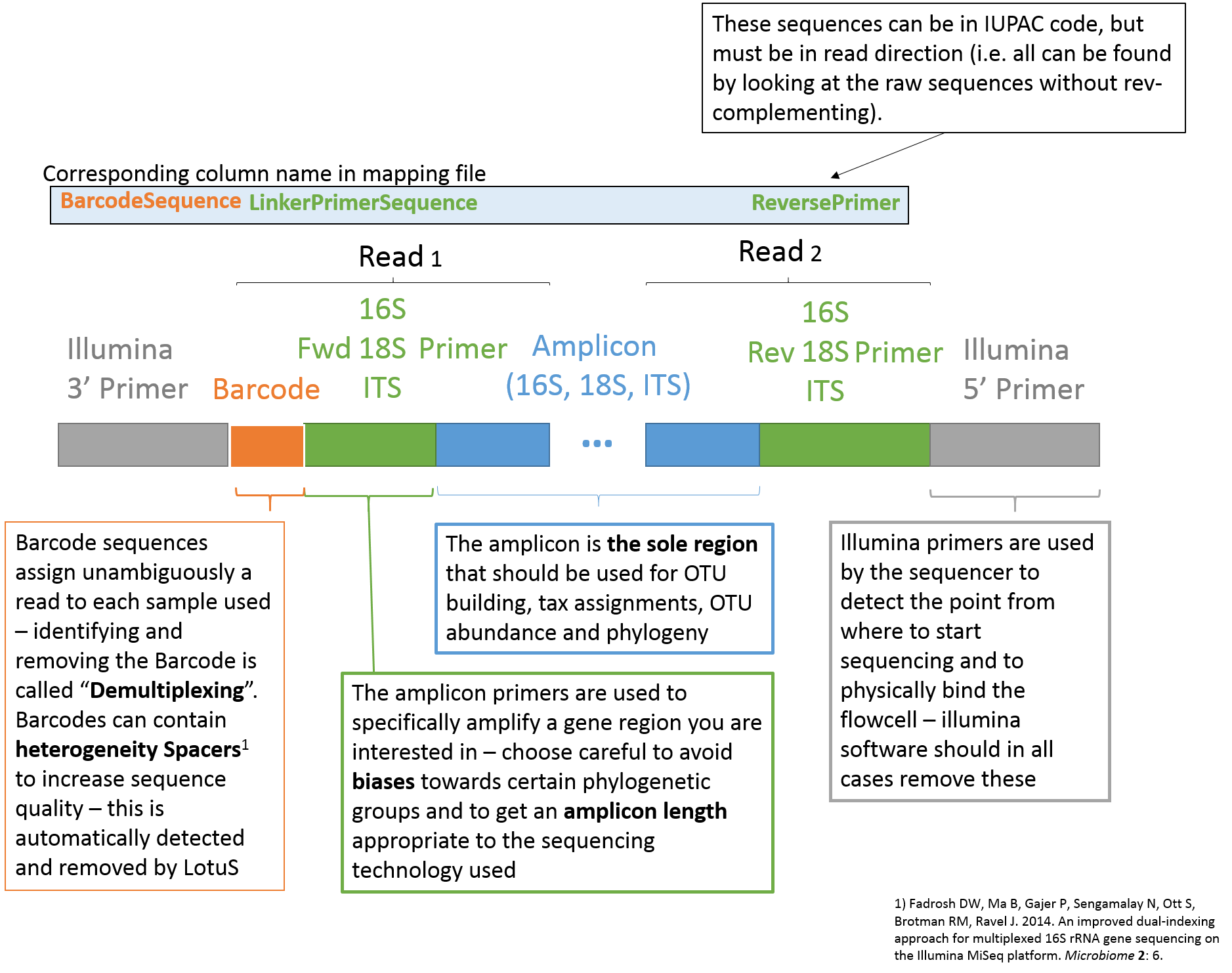
Mapping file configuration
The mapping file, specified via the "-map" or "-m" argument to LotuS2, is used within the pipeline to demultiplex fasta + qual or fastq files, by using the program sdm (included in LotuS2). The first line in the mapping file is the header and has to start with "#". Entries have to be tab separated and the mapping file should be stored as text file. The number and names of columns are not limited (this will be exported to the .biom file) and only columns with column names in the table below will be used for processing the read files. Thus, the desired subparts of the pipeline are activated by having these column names in the header of the mapping file (see also Usage examples).
"automap.pl" creates automatically LotuS2 compatible maps from input dirs, if the run is already demultiplexed. Use perl automapl.pl to get instructions on how to use this helper script.
| Column name | Function |
|---|---|
| SampleID | Sample Identifier, has to be unique for each Barcode. |
| BarcodeSequence | The Barcode (MID) tag assigned to each sample. |
| Barcode2ndPair | in case of dual indexed reads, use this column to specify the BC on the 2nd read pair |
| ForwardPrimer | Sequence used for 16S amplification, usually (unless paired end mode) is after the Barcode. Can contain IUPAC redundant nucleotides. The old tag LinkerPrimerSequence also works for this option. |
| ReversePrimer | Reverse Primer Sequence (IUPAC code) |
| fastqFile | Gives relative location of fastq file, such that [-i][fastqFile] gives the absolute path to fastq file |
| fnaFile | see fastqFile. However, fasta formated file instead of fastq format |
| qualFile | see fastqFile. However, quality file corresponding to fasta file instead of fastq format |
| SampleIDinHead | ID in header of fasta/fastq file, that identifies Sample //This replaces Barcode (MID) scanning\\ |
| MIDfqFile | extra fastq file that contains only MIDs (equivalent to LotuS2 script option "-barcode"). Requires paired reads. |
| CombineSamples | Option to combine samples, that may be distributed across several files. A new tag is set here and all samples with this specific tag will be merged into a new tag with tag as SampleID. Note that SampleID's themselves have still to be unique. |
| SequencingRun | If you use dada2 with multiple sequencing runs, in principle you need to run dada2 separately for each run. If so, please include this information in the mapping file under the "SequencingRun" column or store the input fastq for each sequencing run under different folders. |
Typically, SampleID, BarcodeSequence and LinkerPrimerSequence are required. If the reverse primer should be identified and removed, ReversePrimer is required. If more than 1 sequencing run was used, it is often easier to demultiplex all fasta / fastq sequences at once. For this purpose LotuS2/sdm offers either the fastqFile column, or in case of fasta+qual input files, the two columns fnaFile and qualFile. Within these columns the relative path towards input files can be specified. Note that this format is very similar to the Qiime mapping file format, but for additional options exclusive to LotuS2.
LotuS2 output
After the LotuS2 run has finished, the output folder specified via the -o option contains the following files and subfolders:
| file/folder | Function |
|---|---|
| OTU.txt | OTU abundance matrix |
| OTU.fna | Fasta formatted extended OTU seed sequences |
| OTUphylo.nwk | Newick formatted phylogenetic tree between sequences |
| hiera_BLAST.txt | OTU taxonomy assignments based on Blastn |
| hiera_RDP.txt | OTU taxonomy assignments based on RDP classifier |
| phyloseq.Rdata | Phyloseq object ready to be loaded in R |
| higherLvl/ | Directory with Species, Genus, Family, Class, Order and Phylum abundance matrices |
| primary/ | Directory with copies of sdm options and (sometimes automatically modified) mapping file |
| LotusLogs/ | Directory with log files from the pipeline runs, various statistics to demultiplexing, clustering, taxonomy assignments and quality assurance steps. |
| LotusLogs/LotuS_runlog.txt | This file tracks overall progression and reports the most basic stats of the single processing steps. |
| LotusLogs/LotuS_progout.txt | The concatenated output of all programs run by the lotus pipelines, useful for finding errors in case the pipeline aborts, or just for curiosity. |
| LotusLogs/LotuS_cmds.txt | All commands executed by the LotuS2 pipeline, for reproducibility. |
| LotusLogs/citations.txt | Citations to all programs used by each specific LotuS2 run, please try to acknowledge this software in case of a publication using LotuS2 |
| LotusLogs/demulti.log.* | Log File for sdm (includes a log file for each single fna/fastq file) |
| LotusLogs/dada2_p_errF.pdf | Error profile plots for the forward reads (dada2 option) |
| ExtraFiles/OTU.MSA.fna | Multiple Alignment of OTU’s |
| ... | Auxilliary |
| LotusLogs/demulti.acceptsPerFile.log | Number and percentage of reads that was accepted per sample - can be used to find samples that consistently had reduced read quality. |
| LotusLogs/demulti.acceptsPerSample | Percentage of reads that was accepted per input file - can be used to find sequencer output file that consistently had reduced read quality. |
| LotusLogs/demulti_lenHist.txt | Length histogram of sequences that passed sdm |
| LotusLogs/demulti_qualHist.txt | Quality histogram of sequences that passed sdm |
| LotusLogs/SeedExtensionStats.txt | Min/Avg/Max stats on Seed sequence length, quality accumulated error and similarity to OTU consensus sequence |
Copyright © All rights reserved | This template is made with Colorlib